中国民间中医医药研究开发协会国际针灸合作委员会
办公地点现在已经搬迁至西城区西直门南小街国英园一号楼824室,
同时为方便大家联系,固定电话已经变更
新号码010―58562339。特此通知。
地址:北京西城区西直门南小街国英园一号楼824室
邮编:100035
电话:010-58562339
传真:010-58562339
邮箱:cngjzj@163.com
网站(点击网址直接链接↓):http://www.cngjzj.com/
博客(点击网址直接链接↓):http://blog.sina.com.cn/cngjzj
交通路线图 (点击观看大图)
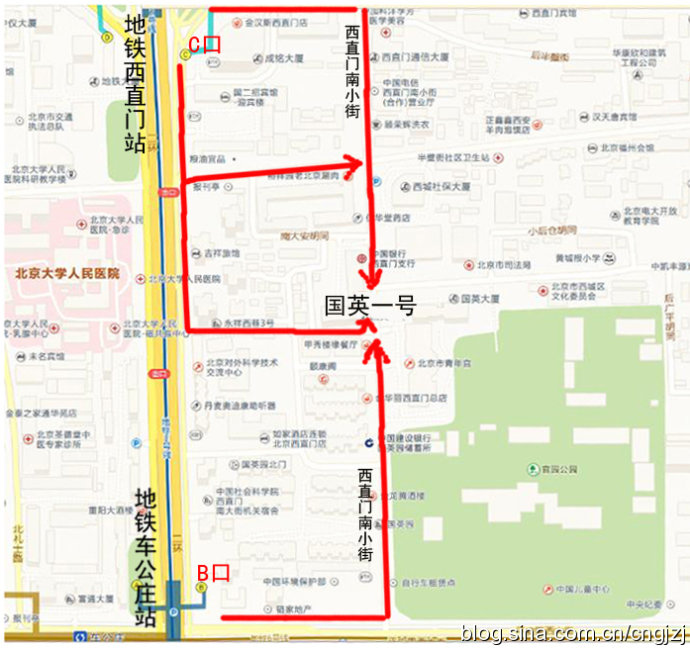
从首都机场乘坐机场专线,在东直门站下车换乘地铁2号线开往西直门方向,在西直门站 C 口出站:
1、沿西直门内大街向东直行100米,右拐到西直门南小街,向南步行到丁字路口即到国英园1号楼楼下。
2、向南直行50米,绕过 国二招宾馆 沿着中大安胡同向东到西直门南小街,向南步行到丁字路口即到国英园1号楼楼下。
从首都机场内乘坐机场直达西单的大巴,在西单站下车,乘坐出租车到西直门南小街国英园1号楼。
公交官园站:107路,运通106路
公交西直门南:387路,44路,800内环,816路,820内环,845路
地铁车公庄:地铁二号线
地铁西直门:地铁二号线
公交车公庄东:107路,118路,701路
公交车公庄北:209路,375路,392路

2010年12月24日
 复制链接
复制链接
 打印
打印
 大 中 小
大 中 小
中药注册管理补充规定
第一条 为体现中医药特色,遵循中医药研究规律,继承传统,鼓励创新,扶持促进中医药和民族医药事业发展,根据《药品注册管理办法》,制定本补充规定。
第二条 中药新药的研制应当符合中医药理论,注重临床实践基础,具有临床应用价值,保证中药的安全有效和质量稳定均一,保障中药材来源的稳定和资源的可持续利用,并应关注对环境保护等因素的影响。涉及濒危野生动植物的应当符合国家有关规定。
第三条 主治病证未在国家批准的中成药【功能主治】中收载的新药,属于《药品注册管理办法》第四十五条第一款第(四)项的范围。
第四条 中药注册申请,应当明确处方组成、药材基原、药材产地与资源状况以及药材前处理(包括炮制)、提取、分离、纯化、制剂等工艺,明确关键工艺参数。
第五条 中药复方制剂应在中医药理论指导下组方,其处方组成包括中药饮片(药材)、提取物、有效部位及有效成分。
如含有无法定标准的中药材,应单独建立质量标准;无法定标准的有效部位和有效成分,应单独建立质量标准,并按照相应的注册分类提供研究资料;中药提取物应建立可控的质量标准,并附于制剂质量标准之后。
第六条 中药复方制剂除提供综述资料、药学研究资料外,应按照本规定第七条、第八条和第九条,对不同类别的要求提供相关的药理毒理和临床试验资料。
第七条 来源于古代经典名方的中药复方制剂,是指目前仍广泛应用、疗效确切、具有明显特色与优势的清代及清代以前医籍所记载的方剂。
(一)该类中药复方制剂的具体目录由国家食品药品监督管理局协助有关部门制定并发布。
(二)符合以下条件的该类中药复方制剂,可仅提供非临床安全性研究资料,并直接申报生产:
1.处方中不含毒性药材或配伍禁忌;
2.处方中药味均有法定标准;
3.生产工艺与传统工艺基本一致;
4.给药途径与古代医籍记载一致,日用饮片量与古代医籍记载相当;
5.功能主治与古代医籍记载一致;
6.适用范围不包括危重症,不涉及孕妇、婴幼儿等特殊用药人群。
(三)该类中药复方制剂的药品说明书中须注明处方及功能主治的具体来源,说明本方剂有长期临床应用基础,并经非临床安全性评价。
(四)该类中药复方制剂不发给新药证书。
第八条 主治为证候的中药复方制剂,是指在中医药理论指导下,用于治疗中医证候的中药复方制剂,包括治疗中医学的病或症状的中药复方制剂。
(一)该类中药复方制剂的处方组成应当符合中医药理论,并具有一定的临床应用基础,功能主治须以中医术语表述。
(二)该类中药复方制剂的处方来源、组方合理性、临床应用情况、功能主治、用法用量等内容由国家食品药品监督管理局药品审评中心组织中医药专家审评。
(三)疗效评价应以中医证候为主。验证证候疗效的临床试验可采取多种设计方法,但应充分说明其科学性,病例数应符合生物统计学要求,临床试验结果应具有生物统计学意义。
(四)具有充分的临床应用资料支持,且生产工艺、用法用量与既往临床应用基本一致的,可仅提供非临床安全性试验资料;临床研究可直接进行Ⅲ期临床试验。
(五)生产工艺、用法用量与既往临床应用不一致的,应提供非临床安全性试验资料和药效学研究资料。药效学研究应采用中医证候的动物模型进行;如缺乏成熟的中医证候动物模型,鼓励进行与药物功能主治相关的主要药效学试验。临床研究应当进行Ⅱ、Ⅲ期临床试验。
(六)该类中药复方制剂的药品说明书【临床试验】项内容重点描述对中医证候的疗效,并可说明对相关疾病的影响。
第九条 主治为病证结合的中药复方制剂中的“病”是指现代医学的疾病,“证”是指中医的证候,其功能用中医专业术语表述、主治以现代医学疾病与中医证候相结合的方式表述。
(一)该类中药复方制剂的处方组成应当符合中医药理论,并具有一定的临床应用基础。
(二)具有充分的临床应用资料支持,且生产工艺、用法用量与既往临床应用基本一致的,可仅提供非临床安全性试验资料;临床研究应当进行Ⅱ、Ⅲ期临床试验。
(三)生产工艺、用法用量与既往临床应用不一致的,应提供非临床安全性试验资料,并根据拟定的功能主治(适应症)进行主要药效学试验。药效学研究一般应采用中医证候的动物模型或疾病模型;如缺乏成熟的中医证候动物模型或疾病模型,可进行与功能(药理作用)相关的主要药效学试验。临床研究应当进行Ⅱ、Ⅲ期临床试验。
第十条 对已上市药品改变剂型但不改变给药途径的注册申请,应提供充分依据说明其科学合理性。应当采用新技术以提高药品的质量和安全性,且与原剂型比较有明显的临床应用优势。
(一)若药材基原、生产工艺(包括药材前处理、提取、分离、纯化等)及工艺参数、制剂处方等有所改变,药用物质基础变化不大,剂型改变对药物的吸收利用影响较小,可根据需要提供药理毒理研究资料,并应进行病例数不少于100对的临床试验,用于多个病证的,每一个主要病证病例数不少于60对。
(二)若药材基原、生产工艺(包括药材前处理、提取、分离、纯化等)及工艺参数、制剂处方等有较大改变,药用物质基础变化较大,或剂型改变对药物的吸收利用影响较大的,应提供相关的药理毒理研究及Ⅱ、Ⅲ期临床试验资料。
(三)缓释、控释制剂应根据普通制剂的人体药代动力学参数及临床实际需要作为其立题依据,临床前研究应当包括缓释、控释制剂与其普通制剂在药学、生物学的对比研究试验资料,临床研究包括人体药代动力学和临床有效性及安全性的对比研究试验资料,以说明此类制剂特殊释放的特点及其优势。
第十一条 仿制药的注册申请,应与被仿制药品的处方组成、药材基原、生产工艺(包括药材前处理、提取、分离、纯化等)及工艺参数、制剂处方保持一致,质量可控性不得低于被仿制药品。如不能确定具体工艺参数、制剂处方等与被仿制药品一致的,应进行对比研究,以保证与被仿制药品质量的一致性,并进行病例数不少于100对的临床试验或人体生物等效性研究。
第十二条 变更药品处方中已有药用要求的辅料的补充申请,如处方中不含毒性药材,辅料的改变对药物的吸收、利用不会产生明显影响,不会引起安全性、有效性的明显改变,则可不提供药理毒理试验资料及临床试验资料;如该辅料的改变对药物的吸收、利用可能产生明显影响,应提供相关的药理毒理试验资料及Ⅱ、Ⅲ期临床试验资料。
第十三条 改变影响药品质量的生产工艺的补充申请,如处方中不含毒性药材,生产工艺的改变不会引起物质基础的改变,对药物的吸收、利用不会产生明显影响,不会引起安全性、有效性的明显改变,则可不提供药理毒理试验资料及临床试验资料;如生产工艺的改变对其物质基础有影响但变化不大,对药物的吸收、利用不会产生明显影响,可不提供药理毒理试验资料,进行病例数不少于100对的临床试验,用于多个病证的,每一个主要病证病例数不少于60对;如生产工艺的改变会引起物质基础的明显改变,或对药物的吸收、利用可能产生明显影响,应提供相关的药理毒理试验资料及Ⅱ、Ⅲ期临床试验资料。
第十四条 需进行药理研究的改变已上市药品剂型、改变生产工艺以及改变给药途径的注册申请,应以原剂型、原生产工艺或原给药途径为对照进行药效学试验(对照可仅设一个高剂量组)。
第十五条 新的有效部位制剂的注册申请,如已有单味制剂上市且功能主治(适应症)基本一致,应与该单味制剂进行非临床及临床对比研究,以说明其优势与特点。
第十六条 非临床安全性试验所用样品,应采用中试或中试以上规模的样品。临床试验所用样品一般应采用生产规模的样品;对于有效成分或有效部位制成的制剂,可采用中试或中试以上规模的样品。
第十七条 处方中含有毒性药材或无法定标准的原料,或非临床安全性试验结果出现明显毒性反应等有临床安全性担忧的中药注册申请,应当进行Ⅰ期临床试验。 第十八条 新药的注册申请,申请人可根据具体情况申请阶段性(Ⅰ期、Ⅱ期、Ⅲ期)临床试验,并可分阶段提供支持相应临床试验疗程的非临床安全性试验资料。
阶段性临床试验完成后,可以按补充申请的方式申请下一阶段的临床试验。
第十九条 临床试验需根据试验目的、科学合理性、可行性等原则选择对照药物。安慰剂的选择应符合伦理学要求,阳性对照药物的选择应有充分的临床证据。对改变已上市药品剂型、改变生产工艺、在已上市药品基础上进行处方加减化裁而功能主治基本一致的中药制剂,需选择该上市药品作为阳性对照药物。
第二十条 临床试验期间,根据研究情况可以调整制剂工艺和规格,若调整后对有效性、安全性可能有影响的,应以补充申请的形式申报,并提供相关的研究资料。
第二十一条 藏药、维药、蒙药等民族药的注册管理参照本规定执行。民族药的研制应符合民族医药理论,其申请生产的企业应具备相应的民族药专业人员、生产条件和能力,其审评应组织相关的民族药方面的专家进行。
第二十二条 本规定自公布之日起施行。
 您现在的位置是:
您现在的位置是:













