�й������ҽҽҩ�о�����Э�������ĺ���ίԱ��
�칫�ص������Ѿ���Ǩ����������ֱ����С�ֹ�Ӣһ��¥824�ң�
ͬʱΪ��������ϵ���̶��绰�Ѿ����
�º���010��58562339���ش�֪ͨ��
��ַ��������������ֱ����С�ֹ�Ӣһ��¥824��
�ʱࣺ100035
�绰��010-58562339
���棺010-58562339
���䣺cngjzj@163.com
��վ�������ֱַ�����ӡ�����http://www.cngjzj.com/
�����������ֱַ�����ӡ�����http://blog.sina.com.cn/cngjzj
��ͨ·��ͼ ������ۿ���ͼ��
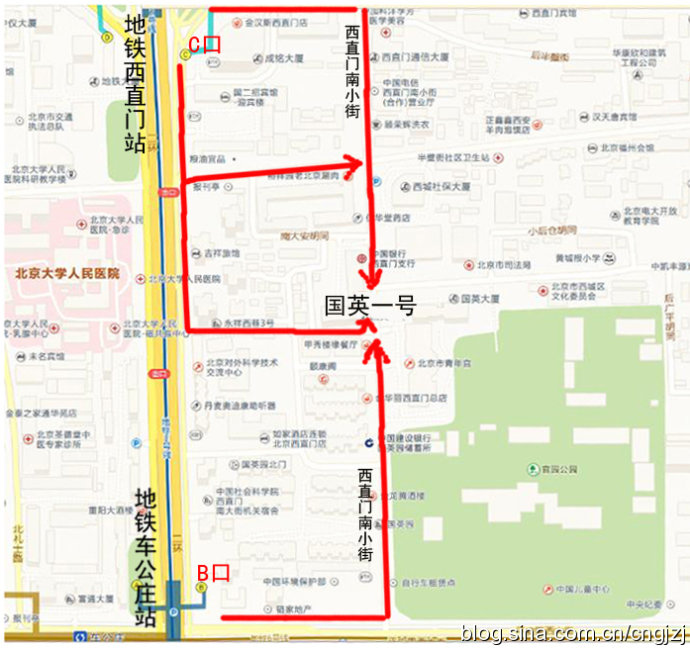
����������������ר�ߣ��ڶ�ֱ��վ�³����˵���2���߿�����ֱ�ŷ�������ֱ��վ C �ڳ�վ��
��������ֱ���ڴ����ֱ��100�ף��ҹյ���ֱ����С�֣����ϲ��е�����·�ڼ�����Ӣ1��¥¥�¡�
2������ֱ��50�ף��ƹ� �����б��� �����д�ͬ����ֱ����С�֣����ϲ��е�����·�ڼ�����Ӣ1��¥¥�¡�
���������ڳ�������ֱ�������Ĵ�ͣ�������վ�³��������������ֱ����С�ֹ�Ӣ1��¥��
������վ��107·����ͨ106·
������ֱ���ϣ�387·��44·��800�ڻ���816·��820�ڻ���845·
��������ׯ������������
������ֱ�ţ�����������
��������ׯ����107·��118·��701·
��������ׯ����209·��375·��392·

2018��12��04��
 ��������
��������
 ��ӡ
��ӡ
 �� �� С
�� �� С
���ߣ�ҩС��
�ĸ↑��40���ڼ䣬�й�ҩƷ�з��������������ı仯��ҩƷת�ü����������־�������Щ�ĸ�ҿ����ġ�
�ĸ↑��40��ҩƷ�з����仯
һ�������о�
��5�������ҹ�����ҩ�з����˺ܴ�̶ȵ�������������2017�꣬���ڻ�ҩ������ҩ�Ĵ���ҩ�걨�����˴���ȵ����ӣ�2017���걨�����ﴴ��ҩ���2016���Ƿ��ﵽ��126.7%��

ͼ1-2-1 2013-2017�괴��ҩ���걨�����Ʒ�ּƣ�
���Ӱе��������ҹ���5�������м��걨�Ĵ���ҩ�����ѷ��֣�������������֪�е��ϵ��ٴ��£��ҹ������о���ת���о���ԱȽϱ��������δ����������д���ǿ��
����ŷ����ҩǿ�����������ҩ�з���������Ҫ��������[1]��
1����ѧ�ͷ�ӯ�����л���
���£��£������������о����������µ�ҩ��е�ʹ����µ��ٴ������ֶΣ�����ҩ����ӣ�ƽ̨�������Ľ�����������Դ����ȫ��Դ�������Կ��壩���о�������Ҫ���Թ��Һ�רҵ��������Ŀ����������о��ɹ��ijɹ���·�Ǽ�����Ȩ��ת�ø���ҽҩ��˾���о���Ա�õ�����Ͷ�ʻ���������������ѧ�Ƽ�����С��˾������ҵ��
2��������������С��˾
���ڹ�˾��ҵ��Ա�����Ĵ��¼����������ڿ�չ��ҩ�з��������о���������Ҫ�������ڷ���Ͷ�ʡ��ɹ��ij�·�DZ�������ҩ��˾����ּ沢��ż��Ҳ��С��˾ƾ�������ƽ̨����������չ�ɴ�˾������������Genetech�������������Խ�500����Ԫ�ļ۸���ʿRoche�ϲ���
3����������ҩ��˾
����һ����һ���Ŀ�ѧ�Һ��̼�����Ա���Լ��侫ϸ�ļ���רҵ�ֹ�����ȡ�����ڹ�����ˮ�ߵ�Э����ʽ������µİе���ڳ����ƽ̨�������д���ҩ����ӵ��з���������µļ���ƽ̨����µĺ��ϵİе��з�����ҩ�����з�����������ҵ��������ʱ��г����ʽ𣬳ɹ��ij�·�Dz�Ʒ���С�
�ӹ�����ҩ������ҵ���ҩ�з������������ҩ�з�����������ҵ����ѧ�����л�����Ҫ���ػ����о���ͬ���أ��ҹ������о�����Ҳ��Ҫ�����ڿ���Ժ�����籱��ҩѧԺ���й�ҩ�ƴ�ѧ������ҩ�ƴ�ѧ���й���ѧԺ�Ϻ�ҩ���о�����ҩ�о������ص�ʵ���ҵȡ�ĿǰҲ���൱һ���ִ�������ҵͨ�����ڿ���Ժ��������о����������ķ�ʽ������ҩ�Ŀ�����������ҵ���������з����ҹ���ҩ�з���;���ͷ�ʽ���Ķ����������Ƕ������뿪�����о���
�ĸ������������ҩ�з��Ƚ����ѣ����������Ȼ��뵽�����ĺ�Ĭ������Ȼ���Ƕ��ڰ����ĺ�Ĭ���ļ�����������һ�����˽⣬�������²�����һֱ���ڡ���˵���Ρ���ˣ�������ĺ�Ĭ������ҩ�з�����ʧ�ܵ����Ҳ������������ˡ�
����ҩƷ����뷨��
�����ܳ�Ϊȫ��������ҩ�۹������䱳��ǿ���ҩƷ��ܻ�����������ʳƷҩƷ�ල�����֣����FDA�������ܲ��ɷֵĹ�ϵ��FDA��ҩƷ�з���ע�ᡢ��ܡ���ͨ���������߷��浼��ĺ������Ƚ��ԣ����ҹ�ҩƷ�����˵���о�Ľ�����塣
�ҹ�ҩƷ����������˺ܶ���ʷ���⣬Ҳ�����˶�θĸ�ع�ÿһ�θĸ�����Ծ����ⱻ��ѹ���������ֳ��ָ��գ��ĸﲢ������ͳ��ס���ˣ���2015��8��18�չ���Ժ�������ڡ��ĸ�ҩƷҽ����е���������ƶȵ���������ҹ���ҩƷҽ����е���������ƶ��ٴν����˴������ظĸ
��һ����ҩ��ؼ�ܷ���ı��
1.��ҩ�����¶���
�ҹ��ԡ���ҩ���Ķ��徭���˶�α��������ҩ���Ķ����1985�ꡰ�ҹ�δ��������ҩƷ������2002�� ��δ�����й������������۵�ҩƷ�����ٵ�2015�������ȷ��Ϊ��δ���й��������������۵�ҩƷ������ҩ�ӡ��й��¡�����ȫ���¡���ת�䣬����������30�ꡣͨ�����ϵظĸ�ҹ��Դ���ҩ����������ҩ������ҩ����������������ʶ��
2.��չҩƷ�������ɳ������ƶȣ�MAH���Ե�
ҩƷ�������ɳ������ƶȵĽ������ҹ�ҩƷ���������ʻ���һ���ĸMAH��̨֮ǰ���ҹ�ʵ�е����������ɺ���������ͳһ����Ĺ���ģʽ��ֻ��������ҵ�ſ�������ҩƷע�ᣬȡ�����յ�ҩƷ���ĺš�MAHʵʩ��ҩƷ�з���ҩƷ����ʵ�ַ��룬ҩƷ�з������Ϳ�����ԱҲӵ��ҩƷ�����ĺţ�ʹ�ҹ�ҩƷ�з���ַ����˱仯��������ҩ�з���ͬʱ��ҩƷȫ�������ڵİ�ȫ�ԡ���Ч�������Ϊ�ϸ��Ҫ���ƶȵ�����������ƶ��ҹ���ҩ�з��Ŀ��ٷ�չ��
3.��ҩ���ٴ���������ĸ�
�ٴ���������ʸ��϶�ʵ�б����������߱��ٴ����������Ļ�����ʳƷҩƷ��ܲ���ָ����վ�ǼDZ����ɽ���ҩƷҽ����еע��������ί�п�չ�ٴ����顣��Ȼȡ�����ٴ��������GCP��ҩƷ�ٴ���������淶���϶��ƶȣ���������ζ�Ž��������ż����෴��ͨ�������ٴ��о���Դ����ǿ�ٴ����������ܵ�Ч�ʣ�ͬʱҲ��һ����֤�ٴ������������
�Ż��ٴ��������������ٴ���������Ĭʾ�����ơ���������ע�������������������Ĺ�ͨ�������ơ�����ҩ���ٴ��������������ҽ����е�ٴ���������ǰ����������Ӧ��ע�������˽��л��鹵ͨ�����������顣�����ٴ����������һ�������ڣ�ʳƷҩƷ��ܲ���δ�����������������Ϊͬ�⣬ע�������˿ɰ����ύ�ķ�����չ�ٴ����顣�����趨����ҩƷ��������ʩ����ѹ���������ڹ涨��ʱ������ɼ�������������ҵ��˵��������������ѹ�����������ڹ����ķ��գ���Ч����ʹ��Դ����Ӧ�á�
���ܾ����ٴ��������ݡ��ھ��������ȡ�õ��ٴ��������ݣ������й�ҩƷҽ����еע�����Ҫ��ģ����������й��걨ע�����롣Ϊʵ�ֹ��ڹ���ҩƷͬ�����������˻�����һ����
����������ҩ��ؼ�ܷ���ı��
������ҩ�����뱻����ҩ������ͬ�Ļ��Գɷ֡����͡���ҩ;�����������õ����ҩƷ�����н���ҽ��֧�������ҩƷ�ɼ��ԣ�����ҽ�Ʒ���ˮƽ����Ҫ���ú����Ч�档�������ձ��ȡ�������º������Ʋ��ص�����ȡ���ڴٽ�����ҩ�з����¡���Ӧ���ϡ��ٴ�ʹ�õȷ�����л���̽����ӡ����Ҫͨ���ƶ��ϸ��ҩƷר����������ԡ�����ר���������ϸ����ơ�ע�ط���ǿ�����ɵ��������á���������������ҵ�������ǿ����������ȴ�ʩ���ٽ�����ҩ�ķ�չ������ͨ������ҩ�����������̡��ƽ�����ҩ���ʹ�á���������Ƥ�顱�ƶȣ�����������FDA���ġ�������ȫ�Ժ���Ч�����۵Ĵ���ҩ��Ǵ���ҩ��ҩƷ������ӡ���飬���ڸ�¼���ַ�������ҩƷ���ר����Ϣ�����ڹ��������ڸ������Ƥ��ɫΪ��ɫ���׳ơ���Ƥ�顱�����ʶ�ʵʩҩƷǿ�����ɵ����ߣ��ٽ�����ҩ��ҵ��չ��
�ĸ↑���������ҹ�����ҩ��ҵȡ���˿��ٷ�չ����ҵ��ģ������������Ʒ�ֲ��Ϸḻ���ڽ�17���ҩƷ������95%���϶��Ƿ���ҩ��Ϊ���Ϲ������Ⱥ�ڵ����彡���������ش��ס���ҲҪ���������ڸ���ԭ���ҹ�����ҩ��ҵ�����ǿ������Сɢ�Ҳ�ľ����Ի����ڣ�ҩƷ��������ϴ�����ҩƷ�г���Ҫ������ԭ��ҩռ�죬����ԭ��ҩ�۸���ߣ��������Ⱥ�ڶԸ���������ҩ������������ҩƷ�ɼ��ԺͿɸ�������ȣ�����һ����࣬������Ҫ�ĸ����ơ�
1.һ�������۹�������ʵ
����2013�꣬����ʳƷҩƷ�ල�����ܾ־�����˿�չ����ҩ��������Чһ�������۹�����2015�꣬ԭ����ʳƷҩƷ�ල�����ַܾ��������ڻ�ѧҩ�����Ч������ʵ�б��������Ĺ��桷��BE������ҩ���ܾ�����չ��Ϊ��������ҵ������չ��ֱ��2016��3�£�����Ժ����������Ժ�칫�����ڿ�չ����ҩ��������Чһ�������۵��������һ�������۹���������ʵʵʩ����ʡ��Ҳ��̳�̨�˷dz�����������֧����ҵ��չһ���Թ����������ʽ���������ͨ��һ��������Ʒ�ָ����б�ɹ����Ŵ���ҩƷһ�������۹�����˳����չ�������ҹ�����ҩ�г��淶���о�����塣
2.�������й�����Ŀ¼����
���й�����Ŀ¼����������һ����Ϊҵ����֪�����֣����й���Ƥ�顷��������FDA�ġ�Orange Book����ͬ�����й���Ƥ�顷�����ҹ�ҩƷ�з�����ͬ�����壬����¼���й����а�ȫ�ԡ���Ч�Ժ������ɿ��Ե�ҩƷ����ȷ���˲α��Ƽ��ͱ��Ƽ�������ʵʱ���¡�
3.�ƶ��������Ƶ�ҩƷĿ¼
2012��-2016�꣬ȫ����631��ԭ��ҩר�����ڣ�������Ϣ���Գơ������Ѷȴ��Լ�һЩ������ҩƷ�г���ģ��С��ԭ���ڷ��Ƹ������ٶȻ�����������ר������ҩ��û����ҵ�������ע�����롣�ڽ������й�����Ŀ¼�����Ļ����ϣ��ƶ��������Ƶ�ҩƷĿ¼��ͨ��������Ŀ¼�ڵ�ҩƷע���������������������Թ���������ҩ��ҵ���з����������з���ע����������ø�����ҩ��ҵͨ������������ʽ��÷����з�Ȩ�棬�ٽ������ٴ����衢��Чȷ�С���Ӧ��ȱ�ķ���ҩ�������У�һ������Խ������ԭ��ҩ�۸�������⣬һ������Խ������ҩƷ���ҹ��̶�ȱ�����⣬�������ҩƷ�Ŀɼ��Ժ�Ӧ����������
2017�꣬ԭ����ʳƷҩƷ�ල�������ܾ���ʽ��Ϊ��������ҩƷע�Ἴ��Э���ᣨICH����Ա����ζ���й���ҩƷ�з���ע����ʻ���·����������ʷ��һ������CFDA����ICH���ҹ�ҩƷ���������ĸ�IJ���Ҳ��Խ���ӿ졣
4.ԭ���ϰ��Ĺ�������
2017��11�£�CFDA�����ġ����ڵ���ԭ��ҩ��ҩ�ø��Ϻ�ҩ����������������Ĺ��桷��ȡ��ҩ�ø�����ֱ�ӽӴ�ҩƷ�İ�װ���Ϻ����������¼��ҩ���ģ�������ԭ��ҩ��ҩ�ø��Ϻ�ҩ����������ҩƷ�Ƽ�ע������ʱһ������������ʵ��ҩƷ��ҩ��ԭ���ϺͰ�װ���Ϲ������������Ƽ�����Ϊ���ģ������Ƽ�����������Ҫ�����ԭ���ϵ�����Ҫ��������ԭ������������Ҫ��Ŀ�ѧ�ԡ�����ԣ����ҩƷԭ���ϺͰ�װ����������ԭ��ҩ�Ĺ��������ƶȵ�ʵʩ��ֹ��ԭ��ҩ���ĺ��ƶȣ�Ҳ����ΪDMF�ƶȵĹ��������ߡ�
������ҩƷ�ı�������
1.��ҩ�����
1999��ʵʩ�ġ���ҩ�����취���У�����ҩʵ�б����ƶȣ�����ҩ�������ڲ���������������ҵ��ע�����롣��ʱ�ҹ�����ҩ�Ķ���Ϊ���ҹ�δ��������ҩƷ����Ҳ����˵��ֻҪ�����ҷ��ƹ���Ʒ�ֵ��й���ҵ�����ܹ������ҩ�����ڡ������Ѿ��������ڵ�Ʒ�֣������µ���ҵ�Ծɻ����ҩ�����ڵ������Ȼ�Dz������ġ�
��ˣ���2002��䲼�ġ�ҩƷע������취�������У�������ˡ���ҩ����ڡ��ĸ��������ڵ���ҩ������ҩƷ�ල�����ֲ���������ҵ�����ͽ��ڡ�
��ҩ�����ֻ��Թ���ҩƷ�����ڽ���ҩƷ�����⣻�����µ���Ӧ֢��2007�桶ҩƷע������취���������ǻ�ҩ1.6�࣬����3.4�࣬��û����������ڡ�
2016�껯ҩע�����ĸ��������ҩ����Ϊ����ҩ��������ҩ������������ͬ���ļ���ڣ�������������Ӧ֢���������������ҩ�з���
��1-2-5 ��ҩ��������ޱ���˵�������������ε���ҩ�⣬�����������ڣ�

2.ҩƷ֪ʶ��Ȩ�����ƶ�
�ҹ���һ����ר��������1984��3��21�հ䲼��ͬ��4��1��ʩ�С���ר������ʵʩ���ڣ��涨��ҩƷ���û�ѧ������õ����ʲ�����ר��Ȩ��1993��ʵʩ�ġ�ר�������н�ҩƷ���û�ѧ������õ����ʴӲ�����ר��Ȩ��������Ҳ����˵��1993��1��1����������뻯����ר��������
ר���ƶ���ҩƷ�з������ź���Ҫ�ĵ�λ��2017��10�¹���Ժ�칫��ӡ������������������ƶȸĸ����ҩƷҽ����е���µ�����������ҩƷ֪ʶ��Ȩ�ı���Ҳ�������Ӧ��Ҫ��
̽������ҩƷר�������ƶȡ�Ϊ����ר��Ȩ�˺Ϸ�Ȩ�棬���ͷ���ҩר����Ȩ���գ���������ҩ��չ��̽������ҩƷ����������ҩƷר�������ƶȡ�
��չҩƷר���������ƶ��Ե㡣ѡ����ҩ��չ�Ե㣬�����ٴ���������������������е�ʱ�䣬�����ʵ�ר����������
����ר��ǿ������ҩƷ�������������ƶȡ��ڹ��������ܵ��ش���в����£���ȡ��ʵʩǿ�����ɵ�ҩƷע�����룬������������������
���ƺ���ʵҩƷ�������ݱ����ƶȡ�ҩƷע�����������ύע������ʱ����ͬʱ�ύ�������ݱ������롣�Դ���ҩ������������ҩƷ����ͯר��ҩ������������������Ʒ�Լ���սר���ɹ�ҩƷע���������ύ������ȡ����δ��¶���������ݺ��������ݣ�����һ�������ݱ����ڡ�
3.��ҩ�����ƶ�
�ҹ������߶����Ӷ���ҩƷ�ֵı�����ʵ����ҩƷ�ֱ����ƶȡ�1991�����Ժ����ҩƷ�ֵı�������������������ƻ����ɹ���Ժ���ƾ�ǣͷ����������������ҽҩ�����ֲμ���ݵġ���ҩƷ�ֱ�����������1992��10��14���ɹ���Ժ�䲼��1993��1��1����ʩ�С�
�ҹ�����ҩƷ�ֱ����ƶ���������������ʩ���Ƕ���ҩƷ�ֵı�����ʩ����ҩƷ�ֱ����ƶȵ�ʵʩ����Ҫ���ã��ٽ�����ҩƷ����������ߣ���������ҩ������ҵ�ĺϷ�Ȩ�棬�ٽ�����ҩ��ҵ�ļ�Լ������ģ���淶������������CFDA���ݿ���ʾ��������2016��6��6�գ����ڹ���308����ҩ����Ʒ�֣�����ҩ�ķ�չ������Ҫ�����塣308����ҩ����Ʒ�������۶���ڵ�Ʒ����46�����������۶��ʮ�ڵ���ҩ����Ʒ����4����������ҩע���������ע���õ��ζ�����Ρ�������ע��Һ�����η���ע��Һ��̵����ע��Һ����Щ��ҩ����Ʒ�ָ�����Ѫ�ܡ��ǿơ����ơ����Ƶȶ����������
Ϊ������ҩƷ�ֱ����ƶȼ�����˩�͡�����Ҫ����������ȷ��棬ԭ����ʳƷҩƷ�ල�������ڡ���ҩƷ�ֱ����������Ļ������ְ䲼�ˡ���ҩƷ�ֱ���ָ��ԭ����ԭ�����ھ������ϸ���Ͻ�һ���淶��ͬʱ������Ϊ�����˴�ͳ��ҽҩ���ƶ��ġ���ҽҩ��������2016��12��15��ͨ������2017��7��1�տ�ʼʵʩ����Ϊ��ҩ����������ϵ����λ������ȷ����ҽҩ���ҹ�ҽҩ��ϵ�е���Ҫλ�ã�Ϊ��ҽҩ��չ�ĸ��������ṩ�˷���֧�š����ң�����ҩ��չս�Թ滮��Ҫ��2016-2030�꣩�����ᵽ��ȫ��ҽҩ������ϵ���ƶ��䲼��ʵʩ��ҽҩ�����о��ƶ��������߷���Ͳ������£��ӿ졶��ҩƷ�ֱ���������������������ҩ���ɱ�����ϵ������������������ʵ����һ�����ƺ��桶��ҽҩ������ʵʩչ����
�����г�֧��
��һ������ҽ��Ŀ¼
2017�����������Դ����ᱣ�ϲ����������µ�ҽ��Ŀ¼��������һ�ι���ҽ��Ŀ¼�ĸ����Ѿ�8��֮�á������ε������������ﱾ�����������ᡢ�������յ����ڵĶ����������ҩȫ�����롣
��1-2-1 2017�����ҽ��Ŀ¼���������з�����ҩƷ�֣���ҩ���֣�

���ڴ���ҩ��˵�������������ش��������������ⲡ�ֵ�ҩƷΪ����һ�����Ʒ�����Խϸߣ��Ի�����˵���Ƹ���̫������ܹ�˳���������ҽ��Ŀ¼���Ի��ߺ���ҵ��˵�������ش�����塣
2017�����ҽ��Ŀ¼���֮��������Դ��ᱣ�ϲ�����ַ����ˡ����ڹ������������ƻ���ҽ�Ʊ��ա����˱��պ���������ҩƷĿ¼��̬���������й���������֪ͨ����������Դ��ᱣ�ϲ�������ҩƷĿ¼��̬�������ƣ����Զ�̬����ҽ��ҩƷĿ¼ʱ���ƽ�����ٴ�����֧�ִ�����ҽ���������������ҽ��ҩƷĿ¼��̬�����ķ�Χ��������ҩƷ��ר��ҩ���Ƕ���Ʒ�֡�Ŀ¼��������Ʒ��Ӧ�ֱ��ȡ�����İ취����Ƚ������������
ҩƷĿ¼��̬�����������ʵʩ�����������е�ҩƷ��ר��ҩ�����ش����ã���Ӱ���ҹ�ҩƷ���г����۸�֣������ƽ��ҹ�ҩƷ�з��Ļ����ԡ�
������ҩƷ�۸�
���ҹ������ڷ���ҩ��������Ч��ԭ��ҩ֮����ڲ�࣬�����ҩƷ�۸��ϲ���ϴ�ʹԭ��ҩר�����ڣ���ҩƷ�۸���Ȼ�Ӹ߲��¡�
��1-2-2ԭ��ҩ���������ҩ�б�۸�Աȣ����֣�

Ŀǰ���ҹ�����ԭ��ר��ҩƷͨ����������̸���γɼ۸����ƽ��ר��ҩƷ�۸ͺ���ҵ����֮���ƽ�⣬����һ���ؼ��㡣1984�꣬��������ͨ����ҩƷ�۸�����ר���ڲ�������������ʽ��������ҩʱ�������������ƽ���˷���ҩ�ʹ���ҩ��ϵ���ȹ����˷���ҩ��չ�����ӳ���ר�������ڣ�Ϊ��ҩ���з��ṩ�㹻�Ķ�����ͨ��������ԼҩƷ�۸��ͬʱ��Ҳ��Ҫ��������ҹ�����ҩ��ˮƽ����С��ԭ��ҩ֮��IJ�࣬��������ƽ��ҩƷ�۸�
����һ�������۹�����չ�����ҹ���ʡ�ж�ͨ��һ�������۵�ҩƷ����ҩƷ�ɹ����������֧��Ҳ�нϴ��չ�������ڴٽ���ԭ��ҩ��������Чһ�µķ���ҩ��ԭ��ҩƽ�Ⱦ�����
��1-2-3 ��ʡ��һ��������Ʒ������֧��

��������ҩ�ش�ר��
�ش���ҩ���ƿƼ��ش�ר����¼�ơ���ҩ�ش�ר�����2008������ʵʩ���ɹ���Ժͳһ�쵼���Ƽ�����ͬ��չ�ĸ�ί��������ͳ��Э������������10������������ɵ��쵼С�飬�Ƽ���Ϊ�鳤��λ�������쵼���ߺͲ���Э����������������ί�������ί���ڱ��ϲ���������Ϊǣͷ��֯���ţ������쵼С��Ҫ������ʵר����֯ʵʩ�������ش���ҩ���ƿƼ��ش�ר���ǵ�ǰ��������ҩ�����Ƶ�����ʩ֮һ��
��1-2-4 �������ش�ר��Ʒ��TOP10����ҵ����ֹ2017��ף�

�ĸ↑��40��ҩƷת�ü��������߱仯
ҩƷ�ļ���ת�÷�Ϊ��ҩ�ļ���ת�ú�ҩƷ��������ת�ã��������������ˡ�����ת�á�������ת�á�������ת�á���MAH�ƶ���ء��ı仯���̡�
һ����һ�Σ�����ת��
���С�ҩƷע������취����2007��7��1�հ䲼��2007��10��1����ʽʩ�У�2009��8��19�գ�ԭ����ʳƷҩƷ��ַܾ�������ӡ����ҩƷ����ת��ע������涨����֪ͨ����ҩƷ����ת��ע������涨�������³Ƹù涨����Ϊ��ҩƷע������취���ĵ��ĸ������ļ���ʽ����ʵʩ��ԭ���ҩƷ����ת�õĹ涨ͬʱ��ֹ��
�ù涨����ҩ����ת�ú�ҩƷ��������ת��ע���걨��������������ȷ�Ĺ涨��
��һ����ҩ����ת��ע���걨������
1.���С���ҩ֤�顷�ģ����ڽ����С���ҩ֤�顷����δȡ��ҩƷ���ĺŵ���ҩ����ת�ã�ת�÷�Ӧ��Ϊ����ҩ֤�顷����������λ��
2.���С���ҩ֤�顷��ȡ��ҩƷ���ĺŵģ�����δ�������ڻ�����ҩ����ڵģ����ڳ��С���ҩ֤�顷��ȡ��ҩƷ���ĺŵ���ҩ����ת�ã�ת�÷�������ҩ֤�顷����������λ�⣬��Ӧ����������ҩƷ���ĺŵ�ҩƷ������ҵ��
������ҩƷ��������ת��ע���걨������
1.���С���ҩ֤�顷����С���ҩ֤�顷��ȡ��ҩƷ���ĺţ�����ҩ������ѽ����ģ����С���ҩ֤�顷����С���ҩ֤�顷��ȡ��ҩƷ���ĺŵ��Ƽ����������ڵĿ�����ҩƷ��������ת�á�
2.δȡ�á���ҩ֤�顷��Ʒ�֣�ת�÷������÷�Ӧ����Ϊ���Ϸ���������ҩƷ������ҵ������һ��������һ��50%���Ϲ�Ȩ��ɷݣ�����˫����ΪͬһҩƷ������ҵ�ع�50%���ϵ��ӹ�˾�ġ�
3.�ѻ�á�����ҩƷע��֤����Ʒ�֣�����������������ԭ����ҩƷע��������ת�ø�����ҩƷ������ҵ��
�ýν�ֹ������ת�ã�������������Ʒ��ת�á�������ȡ�á���ҩ֤�顷����ҩ����ת�û�ҩƷ��������ת�ý����ף�����δ��á���ҩ֤�顷��ҩƷ��������ת�õ���������ϸ�
������ҩƷ����ת��ע������涨�������֣�
�ڶ�����ҩ����ת��ע���걨������
������������������֮һ�ģ���������ҩ����ڽ���ǰ�����ҩ����ת�õ�ע�����룺
��һ�����С���ҩ֤�顷�ģ�
���������С���ҩ֤�顷��ȡ��ҩƷ���ĺŵġ�
���ڽ����С���ҩ֤�顷����δ������ҩ����ڵ��Ƽ�����С���ҩ֤�顷��ԭ��ҩ���ԡ���ҩ֤�顷�˷�֮����Ӧ���ڰ��ա�ҩƷע������취����������Ӧ�Ƽ���ע������������ļ���ڽ���ǰ�����ҩ����ת�õ����롣
��������ҩ����ת�õ�ת�÷������÷�Ӧ��ǩ��ת�ú�ͬ��
���ڽ����С���ҩ֤�顷����δȡ��ҩƷ���ĺŵ���ҩ����ת�ã�ת�÷�Ӧ��Ϊ����ҩ֤�顷����������λ��
���ڳ��С���ҩ֤�顷��ȡ��ҩƷ���ĺŵ���ҩ����ת�ã�ת�÷�������ҩ֤�顷����������λ�⣬��Ӧ����������ҩƷ���ĺŵ�ҩƷ������ҵ��
������ת�÷�Ӧ����ת��Ʒ�ֵ��������պ�����������ؼ�������ȫ��ת�ø����÷�����ָ�����÷����Ƴ������ϸ������3���������ŵ���Ʒ��
��������ҩ����ת�����룬�������ҩƷ�������������ڿ��ư�ȫ�Է��յı����Ӧ��������صĹ涨�ͼ���ָ��ԭ������о����о�������ͬ�걨����һ���ύ��
�ڰ�����ҩ����ת��ע����������֮�������÷�Ӧ���������ת�÷�ԭҩƷ��֤���ļ����������й�Ҫ������ҩƷ������Ӧ���͢����ٴ�����Ⱥ���������
������ҩƷ��������ת��ע���걨������
�ھ���������������֮һ�ģ���������ҩƷ��������ת�ã�
��һ�����С���ҩ֤�顷����С���ҩ֤�顷��ȡ��ҩƷ���ĺţ�����ҩ������ѽ����ģ�
���С���ҩ֤�顷����С���ҩ֤�顷��ȡ��ҩƷ���ĺŵ��Ƽ����������ڵģ�
�����С���ҩ֤�顷����δ������ҩ����ڵ��Ƽ�����С���ҩ֤�顷�������ڵ�ԭ��ҩ���ԡ���ҩ֤�顷�˷�֮���𣬰��ա�ҩƷע������취����������Ӧ�Ƽ���ע������������ļ�����ѽ����ġ�
������δȡ�á���ҩ֤�顷��Ʒ�֣�ת�÷������÷�Ӧ����Ϊ���Ϸ���������ҩƷ������ҵ������һ��������һ��50%���Ϲ�Ȩ��ɷݣ�����˫����ΪͬһҩƷ������ҵ�ع�50%���ϵ��ӹ�˾�ġ�
�������ѻ�á�����ҩƷע��֤����Ʒ�֣�����������������ԭ����ҩƷע��������ת�ø�����ҩƷ������ҵ��
��ʮ��ҩƷ��������ת�õ�ת�÷������÷�Ӧ��ǩ��ת�ú�ͬ��
��ʮһ��ת�÷�Ӧ�������漰��ҩƷ�Ĵ������������ա���������ȫ�����Ϻͼ���ת�ø����÷���ָ�����÷������Ʒ���ơ���ģ�Ŵ���������ղ�����֤ʵʩ�Լ��������ȸ�����������Ƴ������ϸ������3���������ŵ���Ʒ�����÷�������ҩƷӦ����ת�÷�������ҩƷ����һ�¡�
��ʮ�������÷���ҩƷ�������������ա���������Ӧ����ת�÷�һ�£���Ӧ����ԭ��ҩ��Դ���������ࡢ�����ͱ������Լ��������պ��ղ�����Ӱ��ҩƷ�����ı仯��
��ʮ�������÷���������ģӦ����ת�÷���������ģ��ƥ�䣬���÷�������ģ�ı仯����ת�÷�ԭ��ģ10����С��ԭ��ģ1/10�ģ�Ӧ�����¶�����������ز���������֤����֤������ͬ�걨����һ���ύ��
�����ڶ��Σ�����ת��
�����°�GMP��֤�����У�����֤����������ߣ�����һ��������ͨ���°�GMP��֤����ҵ��˵������������������Ϊ�˹���ҩƷ������������ҵ���У�֧���з����������������ͨ��ԭ��ҩ���Ƽ�����ҩ�ĺ��г�ҩ����ҵ֮������������ϣ�֧����ҵ��չ�沢���顢��Դ���ϣ�ʵ�ֹ�ģ������Լ����Ӫ����߲�ҵ���жȣ�2012��12��21�գ�����ʳƷҩƷ�ල�����֡����ҷ�չ�ĸ�ί����ҵ����Ϣ���������������Ϸ��������ڼӿ�ʵʩ����ҩƷ�������������淶�ٽ�ҽҩ��ҵ�����й������֪ͨ������ʳҩ�ల[2012]376�ţ�����ҩƷ����ת������µ�Ҫ��
1.����ҵ�沢�������ҵ�����ڲ��Ż���Դ���ö�������ҩƷ����ת��ע������룬��һ��������������ٶȣ���ʡ��ҩƷ�ල�������Ž��м��������������ֳ�����Լ�������֤��ϵ��ˡ�����Ҫ��ģ�������ҩƷ�ල����������������
2.ҩƷ������ҵ��������ȫ���ּ�����������ģ��ɽ�������ҩƷ�����ڹ涨������ת�ø���ͨ������ҩƷGMP��֤����ҵ����һ�����͵�ҩƷ����������һ����ת�ø�һ����ҵ��
3.ע�������ҩƷ������ҵӦ��2014��12��31��ǰ���������ҩƷ������ҵӦ��2016��12��31��ǰ������Ҫ�����ҩƷ����ת��ע������룬ͬʱ����ע����ӦҩƷ�������ɺ�ҩƷ���ĺš�����ҩƷ�ල�������Ų�����������
���ļ��ķ����������ڹ�����ҵ����ҩƷ����ת�ã�������ͬһ�����������ߵ�ҩƷ�������������������������ֳ�����Ȩ���·���ʡ��ҩƷ�ල�������š�
2013��2��22�գ�����ʳƷҩƷ�ල�����ַ�������������ʵʩ����ҩƷ�������������淶������ҩƷ����ת���й������֪ͨ������ʳҩ��ע[2013]38�ţ�����ʵʩ����ҩƷ�������������淶��ҩƷGMP��������ҩƷ������ҵ�����Ǩ���沢�������漰��ҩƷ����ת���й�����������˵����
һ�������������εģ�������ҩƷ����ת�ã�
��һ��ҩƷ������ҵ�����Ǩ�沢�������Ǩ�ģ�ԭַҩƷ������ҵ��ҩƷ����������ת������ַҩƷ������ҵ��
�������沢������ҩƷ������ҵһ��������һ��50�����Ϲ�Ȩ��ɷݵģ�����˫����Ϊͬһ��ҵ�ع�50%���Ϲ�Ȩ��ɷݵ�ҩƷ������ҵ��˫���ɽ���ҩƷ����ת�á�
����������ȫ���ּ������������ҩƷ������ҵ���ɽ���ӦƷ����������ת�ø���ͨ������ҩƷGMP��֤����ҵ����ͬһ��������Ʒ����������������һ����ת�ø�һ��ҩƷ������ҵ������ԭ��ҩGMP����ģ���ӦҩƷƷ�ֿɽ��м���ת�ã�ת�뷽����ת�ú��ٽ�������ҩƷGMP��֤��
ע�������ҩƷ������ҵӦ��2014��12��31��ǰ���������ҩƷ������ҵӦ��2016��12��31��ǰ������Ҫ�����ҩƷ����ת��ע�����룬����ҩƷ�ල�������Ų���������
��֪ͨ����ע�������ҩƷ�ͷ���ҩƷ�ļ���ת��ע�����������˲�ͬ��ת�����ޡ�
2014��12��8�գ�����ʳƷҩƷ��ܹ����ְܾ칫������������ע�������ҩƷ����ת���й������֪ͨ��ʳҩ���ҩ���ܡ�2014��203�ţ��ٴ�ǿ����ע�������ҩƷ����ת��ʱ�����⣬��2015��1��1������ȷ�˶��Ѿ�������ע�������ҩƷ����ת�õ�ע�����룬������ʡ��ҩƷ�ල�������Ž�������������δ��������ҩƷ����ԭҩƷ����ת��Ҫ��CFDA�����������������ҩƷ������ҵӦ��2016��12��31����Ȼ����ҩƷ����ת�á�
����ҩƷ�������ɳ������ƶȣ�MAH����ʵʩ
��һ��MAH�ƶȹ������Ե�
2015��8��18�գ�����Ժ���������ڸĸ�ҩƷҽ����е���������ƶȵ��������������2015��44�ţ���������������չҩƷ�������ɳ������ƶ��Ե㡣����ҩƷ�з������Ϳ�����Ա����ע����ҩ����ת�ø���ҵ����ʱ��ֻ����������ҵ�ֳ����պ˲�Ͳ�Ʒ���飬�����ظ�����ҩƷ�����������Ե㹤�������շ�������ȡ����Ȩ��չ��
2016��6��6�գ�����Ժ�칫������������ӡ��ҩƷ�������ɳ������ƶ��Ե㷽����֪ͨ�������췢��2016��41�ţ������ݡ�ȫ�����������᳣��ίԱ�������Ȩ����Ժ�ڲ��ֵط���չҩƷ�������ɳ������ƶ��Ե���й�����ľ��������ڱ�������ӱ����Ϻ������ա��㽭��������ɽ�����㶫���Ĵ���10��ʡ���У���չҩƷ�������ɳ������ƶ��Ե㡣ʵʩ��2018��11��4�ա���־���ҹ�ҩƷ�������ɳ������ƶȿ�ʼ������
2017��10��8�գ��й�����칫��������Ժ�칫��ӡ������������������ƶȸĸ����ҩƷҽ����е���µ��������
����ʮ�����ƶ��������ɳ������ƶ�ȫ��ʵʩ����ʱ�ܽ�ҩƷ�������ɳ������ƶ��Ե㾭�飬�ƶ���ҩƷ������������������ȫ���ƿ�������ҽ����е�з������Ϳ�����Ա����ҽ����е�������ɡ�
����ʮ������ʵ�������ɳ����˷������Ρ�ҩƷ�������ɳ��������ҩƷ�ٴ�ǰ�о����ٴ����顢�������졢�������͡�������Ӧ����ȳе�ȫ���������Σ�ȷ���ύ���о����Ϻ��ٴ�����������ʵ�����������ݣ�ȷ������������������һ�����������̳����Ϲ棬ȷ�����۵ĸ�����ҩƷ���걨��Ʒ����һ�£�ȷ��������ҩƷ���г����о�����ʱ���淢���IJ�����Ӧ���������������������Ľ���ʩ��
ҽ����е�������ɳ��������ҽ����е��ƿ������ٴ����顢�������졢�������͡������¼�����ȳе�ȫ���������Σ�ȷ���ύ���о����Ϻ��ٴ�����������ʵ�����������ݣ�ȷ��������ҽ����е���г����о�����ʱ���淢���IJ����¼����������������������Ľ���ʩ��
��ҩƷҽ����е�������ɳ�����ί�н����з����ٴ����顢�������졢�������͵���ҵ���������ˣ���е����ɷ���涨�����κ�Э��Լ�������Ρ�
����ʮ�ģ������������ɳ�����ֱ�ӱ��治����Ӧ�Ͳ����¼��ƶȡ��������ɳ����˳е�������Ӧ�Ͳ����¼�������������Σ��������������ڱ���ģ��������ϳʹ���ʳƷҩƷ��ܲ���Ӧ�Ա���IJ�����Ӧ�Ͳ����¼����е�����������������������ɳ����˲�ȡ��ͣ���ۡ��ٻء������������Ƶȴ�ʩ��
ҩƷ�������ɳ������ƶȣ�MAH���ĺ���������ҩƷ���ĺź������������룬�����Ե��ҩƷ�з������Ϳ�����Աȡ��ҩƷ�����ĺţ����Ҷ�ҩƷ�����е���Ӧ�����Ρ�MAH��̨֮ǰ�����ǹ���ʵ�е����������ɺ���������ͳһ����Ĺ���ģʽ��ֻ��������ҵ�ſ�������ҩƷע�ᣬȡ�����յ�ҩƷ���ĺš�����ҩƷע�����������ɡ�����ģʽ�������ڹ������£������ڱ���ҩƷ��Ӧ�����������Ƶ�ˮƽ�ظ����衣��չҩƷ�������ɳ������ƶ��Ե㹤�������ڹ���ҩƷ���¡�����ҩƷ����������Ҫ���塣
��1-2-6��ʡ��ҩƷ�������ɳ������ƶ�ʵʩ����

������MAH�ƶ�ȡ�õĽ��Գ�Ч
����ʳƷҩƷ�ල�����ܾ�ͳ���˽���2016��12��25�ո��Ե�ʡ��ҩƷ�������ɳ������Ե�Ʒ���걨��������±���ʾ��
��1-2-7 ҩƷ�������ɳ������Ե�Ʒ���걨�����������2016��12��25�գ�

2016��12��23�գ���ɽ��ʡ��³��ҩ����˾�з��Ŀ�������ҩ�������ἰƬ��������ʳƷҩƷ����ܾ���ȡ�ó������ĺţ���Ϊ�ҹ���ҩƷ�������ɳ������ƶ��Ե�Ʒ�֡�
2017��3��27�գ��㽭ҽҩ�ɷ�����˾�²���ҩ����ƻ������ŵɳ��ԭ��ҩ���佺�Ҽ�������ʳƷҩƷ����ܾ���ȡ����ҩƷ�������ɳ������ƶ��Ե�Ʒ�֣��㽭ҽҩ�ɷ�����˾�²���ҩ��Ϊ��Ʒ�������ɳ����ˡ������ҹ�ʵʩҩƷ�������ɳ������ƶ��Ե㹤��������������������ҩ
2017��8�£��㽭����ҩҵ���Źɷ�����˾�걨�ĵ����ڷ�Һ������ʳƷҩƷ����ܾ���ȡ������ҩ����������ͬʱ�ù�˾���ҩƷ�������ɳ����ˡ������ҹ�ʵʩҩƷ�������ɳ������ƶ��Ե㹤�����������ŵ�����ҩ��ҩ�������ɳ������ĺš���־����ҩ��ҩ���������ɳ������ƶ��Ե㹤�������˿�ϲ��һ����
2018��6�£��Ϻ���������ҩ��������˾��Ϊ�����һ���������ɳ������ĺŵ�ҩƷ�з�������2016��9�£���������MAH���������������ʳƷҩƷ�ල�������ύ����³˾���ƾ�Ƭ����ͨƬ���������롣������Ʒ�����Ϻ���ʳƷҩƷ�ල�����ֹ����������μ�MAH�Ե�Ʒ�֡�����������ί�и���������������ҩ����˾������
������
������Դ�ڼ�������ġ��й�ҽҩ�з�40������ݡ�һ�顣���д�©����ӭ����ĩ���������������ۡ�
 �����ڵ�λ���ǣ�
�����ڵ�λ���ǣ�













